In our last post, we discussed the structural properties that enable Helicons to enter cells and engage challenging targets. Now, let’s turn to the platform that makes Helicon discovery possible. The vast chemical landscape that Helicons can access offers great potential across a wide range of therapeutic applications if we can create, explore, and optimize in this space efficiently.
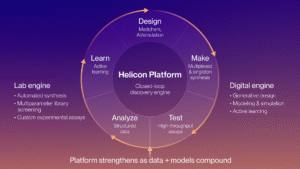
In this post, we’ll break down how our discovery platform delivers on this promise across three key pillars: the chemistry that enables Helicons to be synthesized readily across countless possible configurations, the experimental data generation that produces large volumes of information in a short timeframe, and the computational capabilities that guide the identification and development of Helicons with the drug-like properties required for clinical success.
Chemistry
At the core of our platform is the chemistry used to constrain peptides in an alpha-helical conformation (for more detail on helices, see the prior post, Why Helicons?). As first described in the original 2000 JACS article on helical peptide stapling, this involves using two or more non-natural amino acids to form crosslinks, or “staples,” that rigidify the peptide in a helical conformation. Helical stability is driven by both the conformational restraint of the staple and the particular chemistry of the stapling amino acids. The original stapling systems contained just all-hydrocarbon chains, but today, Parabilis has an internal library of many hundreds of staple types, which cover a diverse range of properties such as polarity, rigidity, and length, and which are formed using a variety of chemical reactions that allow us to incorporate multiple staples and to control their position and geometry.
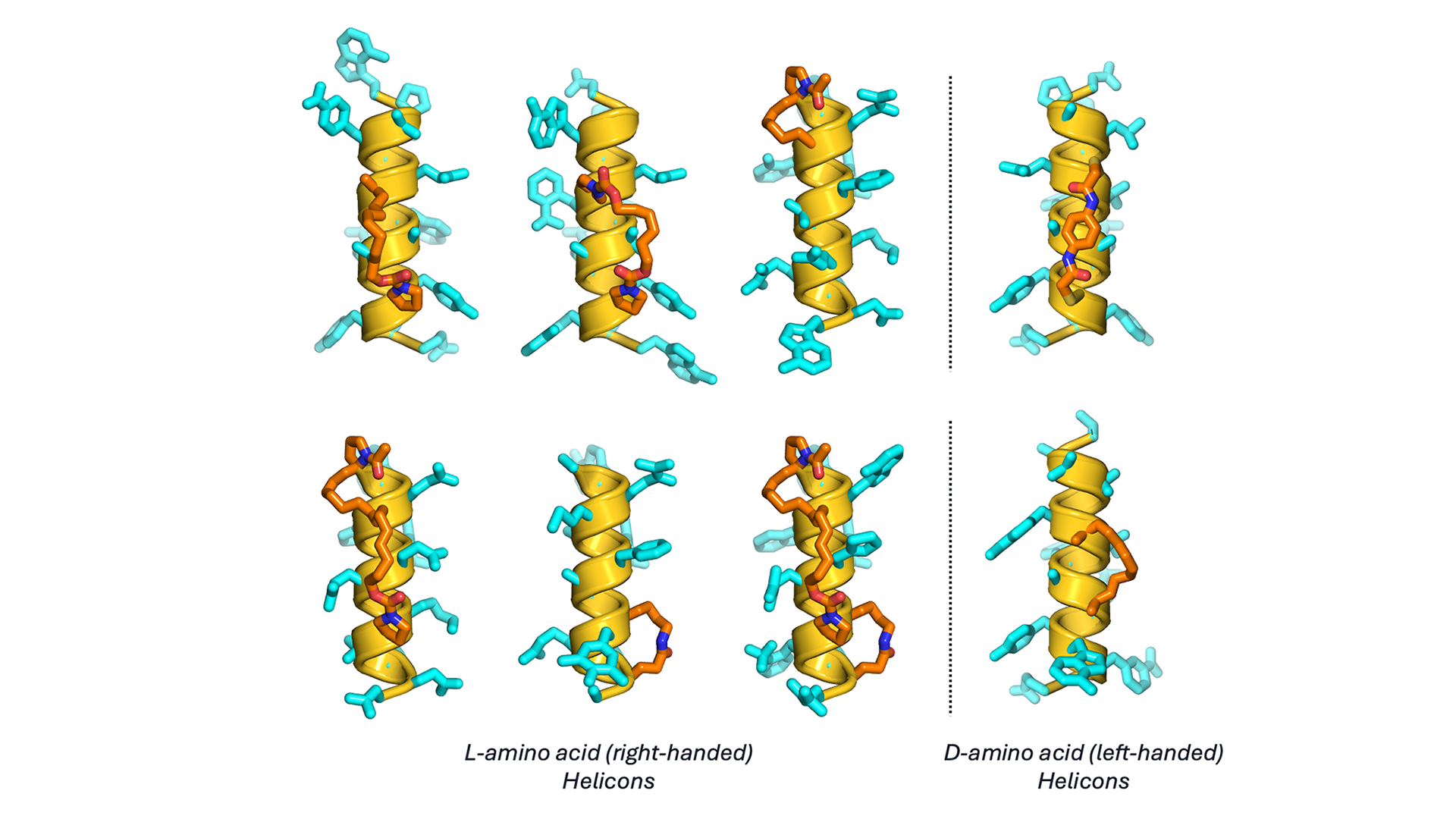
Figure 1. Various peptide staples utilized at Parabilis
Our chemistry platform also contains a second critical dimension of diversity: the library of amino acids, linkers, and ligands used to confer all the drug-like properties our Helicons require to be effective therapeutics. We have more than 1,000 non-natural amino acids (both L– and D-configured amino acids forming inverted helical handedness Helicons) in our internal library, along with dozens of linkers that in turn can be connected to dozens of ligands to recruit E3 ligases, bind essential targets, or deliver radioisotopes. Because of the robust and modular nature of peptide synthesis, these reagents can be readily combined combinatorially to form a truly astronomical number of possible Helicons. If we consider a 15-amino acid peptide containing two stapling amino acids and thirteen non-stapling amino acids, and consider just 100 possible staple types at fixed positions in the peptide and 1,000 possible side-chains at the remaining positions (both significant under-estimates of our current library), we can access 100*100013 = 1041 unique Helicons, any one of which can be made within days of being designed using our highly automated and optimized synthesis and purification platforms. Adding linker and ligand diversity, such as for a targeted degrader or other induced proximity modalities, further increases this diversity by many orders of magnitude. Access to this kind of diversity creates a sufficiently large chemical space to very finely tune the properties of our Helicons to match exactly the profile needed for a given therapeutic application.

Figure 2. The image illustrates more than 1,000 non-canonical amino acid monomers and their chemical structure relatedness. Each node is an amino acid colored by its nearest neighbor and is connected by edges signifying Tanimoto similarity >0.5.
Experimental data generation
Rapid access to such astronomical diversity creates a new challenge: what molecules should we actually make? How do we navigate the universe of possibilities? This challenge brings us to the second pillar of our platform: experimental data generation. Our early focus in this area was centered on hit discovery, since it was essential for us to be able to find hits for any target we thought was worth pursuing. Unlike small molecules or antibodies, Helicons aren’t available in off-the-shelf libraries, so to enable de novo hit discovery, we built a high-diversity screening method based on phage display. Our approach combines on-phage stapling with a highly parallel, direct-to-NGS format that rapidly generates detailed binding data across targets, counter-targets, doses, and conditions. For more details, you can read our paper on the method here. We have now expanded the diversity of this platform by building an mRNA display screening platform for Helicons, which will be the topic of future posts in this blog.
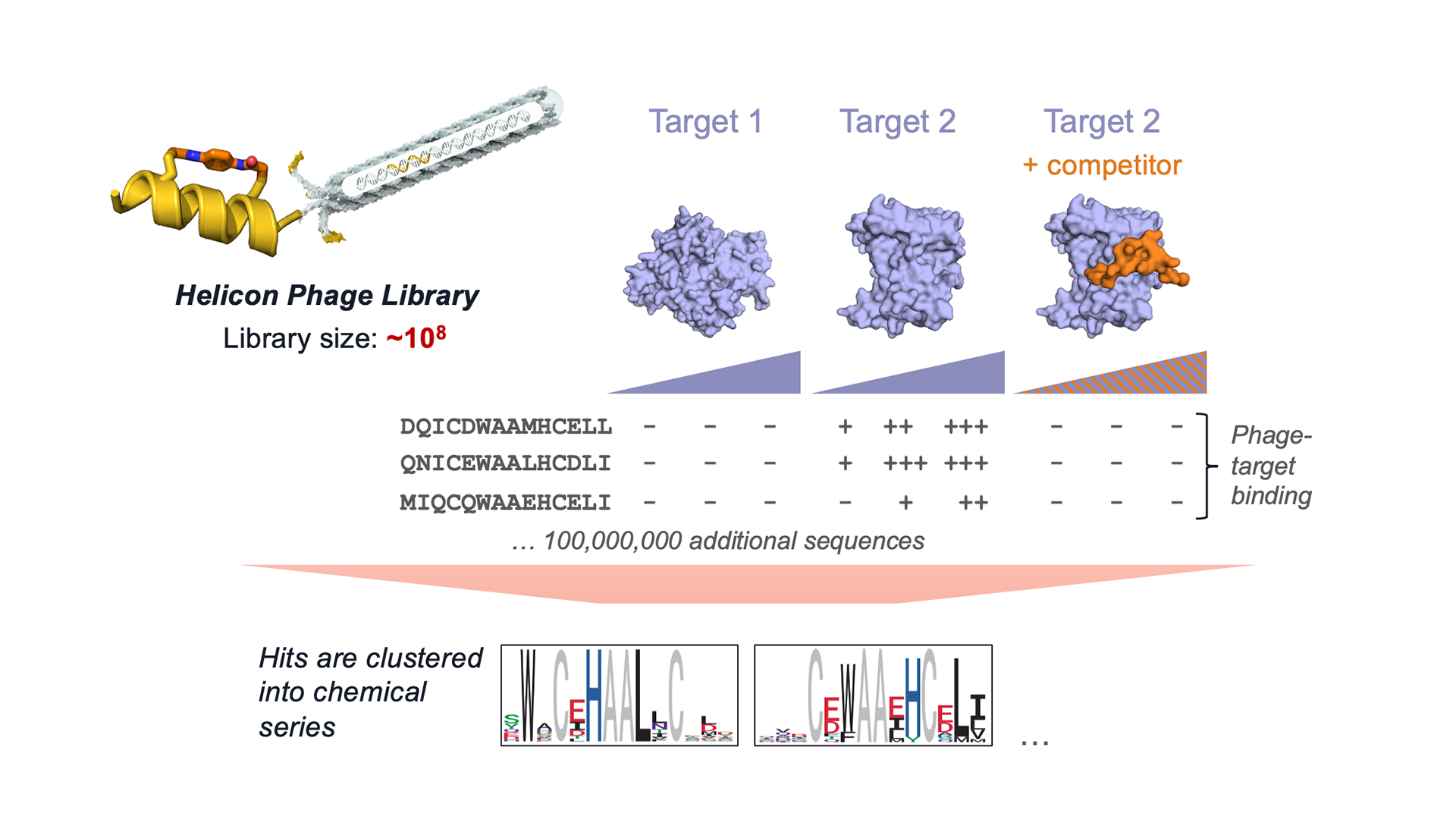
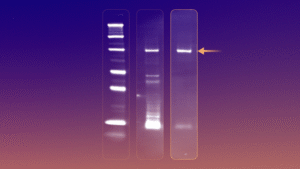
Figure 3. Our highly parallelized Helicon phage screening platform
Once hits have been discovered, there are a range of key initial properties that must be optimized, such as affinity, stability, solubility, and (crucially) cellular penetration. Because these properties can’t be reliably measured on Helicons fused to phage, we built a complementary set of solution-based, tag-free methods that are compatible with cellular assays while still enabling exploration of significant molecular diversity. Many of these methods rely on liquid chromatography-mass spectrometry (LC/MS) readouts to enable “multiplexing” – running mixtures of Helicons in a single well to enable significantly more data generation than would be possible with conventional assays. This includes an assay we built to quantify cytosolic exposure in cells, and to distinguish it from simple cell association, since many peptides can appear to be associated with cells without actually being accessible to targets within the cytoplasm or nucleus. We’ll cover these mass spectrometry–based, multiplexed assays in more detail in future blog posts.
Computational capabilities
Our computational discovery platform closes the loop between the vast chemical space of Helicons and the rich experimental datasets we generate. Starting from either crystal structures or 3D predicted structures, we use “Helisite” prediction from our custom Helisite Finder algorithm, which systematically evaluates a protein structure, including dynamics, to identify binding sites that are compatible with Helicon engagement. Many proteins for which we identify Helisites lack the well-defined pockets required for small-molecule drugs. Thus far, we have achieved very high positive and negative predictive rates, respectively, in many prospectively tested screening and computational de novo design campaigns with the Helisite Finder application. Using these predicted Helisites, we run computational de novo design campaigns in parallel with our phage and mRNA display platforms, with a particular focus on discovering D-Helicons, which expand the chemical space for which traditional display methods cannot directly probe. The computational de novo method also has the advantage of working in the non-natural amino acid space from the beginning for both L- and D-Helicons. Using predicted protein structures alone, we’ve designed novel Helicons against previously unaddressed binding sites – bringing together AI and physics-based methods to go straight from structure to synthesis.
Across the broader design–make–test–analyze cycle from hit ID to late lead optimization, we pair large-scale molecular simulations with machine learning (ML) models trained on a dataset of more than 300,000 Helicons. The ML models result from a bespoke ML pipeline that is custom to Helicons and coupled with large in-house, high-quality data, and have proved impactful to shorten design cycles. Our ML model pipeline has enabled robust global models of key drug properties such as logD, solubility, and cytosolic penetration, to the point that we are often able to replace routine experimental assays with in silico predictions. In addition, we have assembled a growing suite of models that capture additional physicochemical properties, binding affinity, cellular activity across a range of assays, and increasingly, in vivo and in vitro ADME behavior. These supervised models are routinely used within our generative AI engine, developed for Helicons, which is used to solve common and vexing multi-parametric optimization (MPO) problems (i.e., identifying Helicon designs that are optimal for two or more usually compensatory properties). We have also developed a rapid AI-based ΔΔG prediction system that approaches the level of accuracy of free energy perturbation (FEP) at roughly 105-fold lower compute times, allowing us to explore larger spaces of Helicon design faster per design cycle. Finally, large-scale molecular dynamics simulation pipelines for ternary complexes have allowed us to extend the design reach of Helicons into heterobifunctional molecules (such as degraders, RIPTACs, induced proximity, and radioligand therapeutics). All of these capabilities are integrated into internal systems that track designs, automate data ingestion, update models, and surface results through analytical dashboards and soon AI-driven agents, ensuring that each experimental cycle feeds directly into smarter, more targeted design in the next round.
We work to evolve rapidly and continuously every single aspect of our platform, both by building new capabilities computationally and in the lab, and by refining those that are in place. The initial proof-of-concept is often just the beginning of creating a useful discovery technology – there is nearly always a sustained period of continual improvement needed to cover the “last mile” of development to make a tool robust and able to make an impact across multiple programs. Ultimately, reliability and repeatability are the true tests of a platform. In our next post, we will zoom out and talk about some of the other strategic roles that our platform plays in our company and mission.
Authored by Donovan Chin, Data Science & Engineering team member, and John McGee, Platform Technology team member